发布时间:2021-03-09来源:供稿:刘兴国
近日,中国科学院广州生物医药与健康研究院刘兴国课题组构建了线粒体DNA缺失综合症(MDS,Mitochondrial DNA depletion syndrome)病人特异性的及其CRISPR/Cas9修复的诱导多能干细胞,进而分化高纯度3D肝类器官和2D肝样细胞作为肝脏疾病的联合模型。该研究发现MDS病人肝细胞对铁沉积导致的铁死亡(一种细胞的死亡方式)更为敏感,阐明线粒体溶酶体互作引发铁死亡的病理,并筛选出N-乙酰半胱氨酸(NAC)作为潜在的候选药物。这项研究揭示了线粒体疾病的全新死亡模式、细胞器互作机制和潜在治疗策略,相关成果于3月8日以A combined model of human iPSC-derived liver organoids and hepatocytes reveals ferroptosis in DGUOK mutant mtDNA depletion syndrome发表在Advanced Science(《先进科学》)上。
线粒体是真核生物细胞中最重要的细胞器之一,除了为细胞提供能量外,它还参与调控细胞代谢、氧化应激、细胞死亡等多种生理活动,在细胞生、老、病、死等各方面发挥重要作用。线粒体DNA缺失综合症是由于维持线粒体DNA合成的核基因突变,线粒体DNA含量严重减少,导致多组织器官功能障碍的重大疾病,受累器官通常有肝脏、脑、肌肉等。病理表型具有组织特异性,目前已发现至少9种基因突变会导致MDS。刘兴国课题组在前期工作中报道了丙戊酸诱发Alpers-Huttenlocher综合征(由POLG突变导致的MDS)肝毒性的机理,并建立了相应的候选药物筛选策略,是首次利用诱导多能干细胞(iPSC)技术建立遗传特异的肝细胞毒理学模型,成为解决临床问题的成功范例(Xingguo Liu*, Hepatology, 2015)。此后,在线粒体疾病方向,刘兴国团队进行了持续深入的研究。
DGUOK是脱氧鸟苷激酶,是线粒体内合成嘌呤核苷酸重要的酶,该基因突变是导致肝脑型MDS最常见的遗传背景因素。患者大多出生后1个月内发病,预后极差,通常一年内死于严重肝衰竭,除了肝移植,没有其他有效的治疗方法。肝脏铁沉积是其重要临床表型,血清学检查也显示血清铁蛋白和转铁蛋白升高。肝脏作为人体内主要的铁贮器官,铁过载时肝脏首当其冲成为铁毒性攻击的主要部位。然而铁在这个疾病中起何种作用?相关研究至今一片空白! DGUOK突变的MDS病人进展如此迅速且严重的肝衰竭病理机制至今尚未解释清楚,更无有效的针对性治疗药物,亟需深入研究,探寻有效治疗手段。
团队为了攻克这一医学难题,将病人皮肤成纤维细胞重编程为iPSC,进行CRISPR/Cas9基因修复,保证了遗传背景的一致性。接着,团队利用高纯度3D肝类器官分化培养技术排除胆管细胞干扰,并结合2D肝样细胞分化技术,建立了一个更为强大可靠的体外肝脏疾病模型。
团队发现病人肝细胞线粒体DNA缺失导致线粒体功能障碍、ATP合成减少、和活性氧(ROS)大增。病人3D肝类器官和2D肝样细胞均对铁沉积导致的铁死亡更敏感。这一铁死亡是线粒体与溶酶体的细胞器互作事件:首先线粒体活性氧激增及谷胱甘肽耗竭,继而核受体共激活因子4(NCOA4)介导铁蛋白在溶酶体中降解,铁蛋白里的铁释放到胞质中,引起脂质过氧化增加,最终导致肝细胞铁死亡。进一步的工作筛选出谷胱甘肽的前体—N-乙酰半胱氨酸(NAC)可以显著抑制病人肝细胞铁死亡。
该研究首次将高纯度3D肝类器官技术应用于遗传性肝病研究,论证了MDS疾病发生铁死亡这一全新病理,揭示了其临床肝脏铁过载的奥秘,并筛选出有效的抑制铁死亡候选药物。更重要的是,不限于MDS,线粒体DNA缺失广泛存在于衰老、退行性疾病和其他遗传性疾病中,所以该发现具有广泛的潜在病理和治疗意义。
本研究与山东大学合作完成,获国家重点研发项目、中科院、国家自然科学基金、广东省和广州市的经费支持。
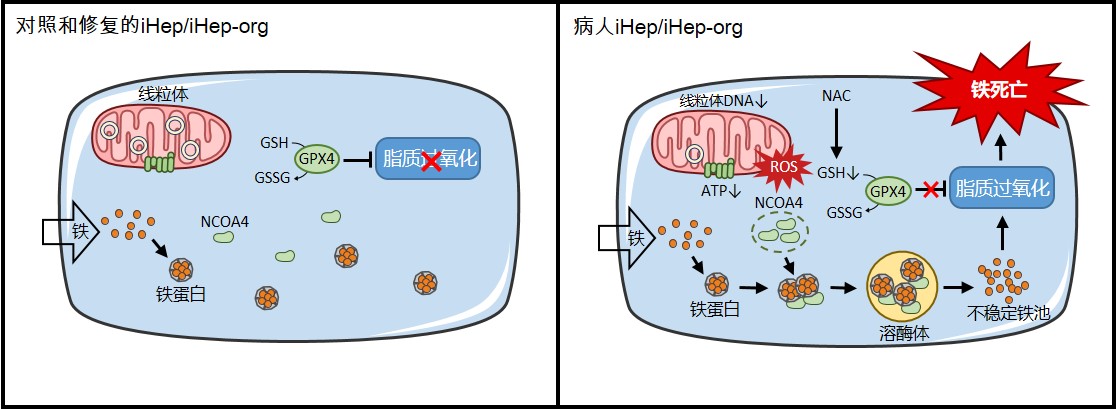
线粒体疾病肝细胞的全新铁死亡模式、细胞器互作机制
附件下载:





